26
2021.03
【全長16S rRNA定序-從屬進階到種的新高度】
原創文章 引用請註明出處
16S 核糖體 RNA (16S ribosomal RNA, 16S rRNA) 為原核生物核醣體小次單元的重要組成,16S rRNA 序列包含保守區以及 9 個高度變異區,稱為 V1-V9。而這些高度變異區域是具有特異性的序列,可做為細菌系統發育和分類的依據。因此,16S rRNA 基因定序已被廣泛用於鑑定各個領域的細菌研究。
全長 16S rRNA 從 V1 到 V9 通常約 1,500 個鹼基對。過去,檢測 16S rRNA 菌相的主要工具是次世代定序 (Next-generation sequencing, NGS)。由於次世代定序的讀長限制,大多數 NGS 技術只能檢測到 16S rRNA 的一部分。例如,Roche-454 定序儀檢測 V1 至 V3 區域,而 Illumina 技術通常定序 V3 和 V4 區域的序列。然而,最近研究發現,只讀一部份的 16S rRNA 去做細菌的註解,會使得某些細菌被偵測到的機率較低,進一步影響統計出來的菌相或者細菌豐度。
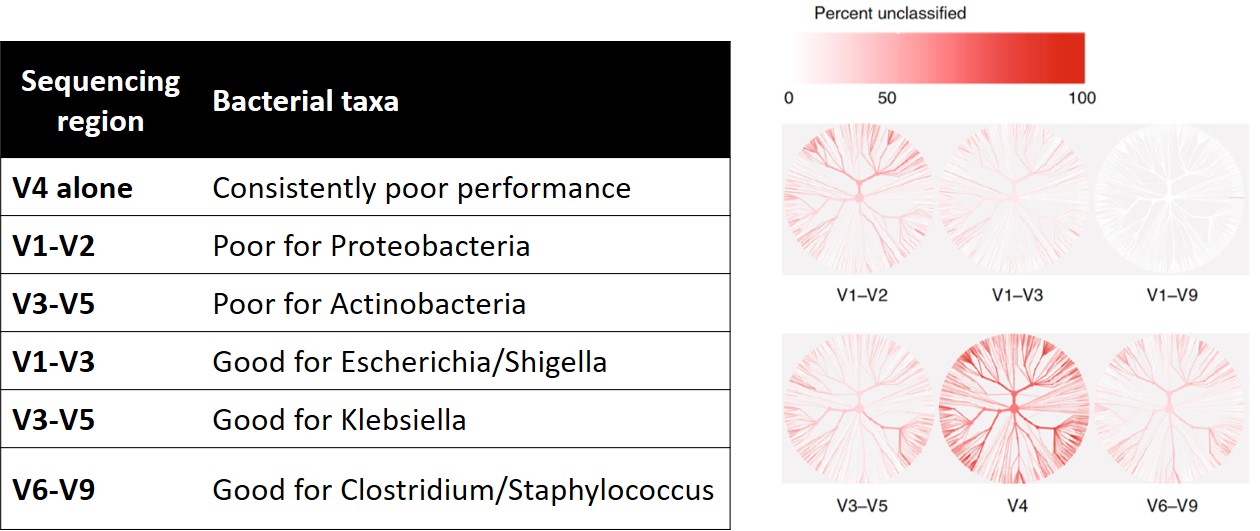
2019 年一篇由 Jethro S. 等人發表在 Nature 的文章中指出,進行菌相分析時,只讀取 16S rRNA 部分區域的序列,可能會使得細菌在分類上效果不好。如下左表顯示[1],當只定序 V1-V2 區域時,對於變形菌門 (Proteobacteria) 的辨識度是差的,只定序 V3-V5 區域時,對於放線菌門 (Actinobacteria) 的辨識度也不好。根據右圖物種階層熱樹圖[1],僅定序 V4 區域時,樹狀圖呈現深紅色分枝,表示未被分類到的細菌百分比很高。當 V1-V9 全部定序時,其樹狀圖顏色接近白色,表示大部分的菌都是有被分類到的。另一方面,只定序部分區域,在細菌分類學分析時,並不是一直都不適用的。例如,大腸桿菌和志賀氏菌的辨別度在只定序 V1-V3 區域時,也有良好的分辨度。總結來說,若需要做菌相分析時,進行 V1 到 V9 全區域的定序,對於微生物菌相分析時,可以提供更良好的分類結果。
第三代定序 (Third-generation sequencing, TGS) 隨著技術越來越成熟,準確度及通量上的提升,近期的研究與應用也越來越多。相較於次世代定序,可以讀取較長片段的三代定序,是作為 16S rRNA V1-V9 全區域定序的好工具。與二代定序相比,三代定序技術可以輕鬆定序超過 20 Kb,足以對全長 16S rRNA 進行定序。尤其是使用 PacBio 技術,定序可以實現長讀長、高準確度,以及平均定序等特性。
在三代定序還未被應用前,16S rRNA 部分區域定序,可以將細菌鑑定到屬的層級。根據研究顯示,分類至屬的層級時,95% 的定序準確度將導致50%的細菌被錯誤分類。而當鑑定的層級要從屬進階到種時,所需的定序準確度要求更高,應將定序準確性提高到至少 98% 以上,在這個準確度的情況下,約還有 50% 的細菌會被錯誤分類 (data from Dr. Zhu Baoli, Institute of Microbiology, CAS)。因此,要定序 16S rRNA V1-V9 全長區域,所需的技術需要讀的長且準。綜合以上所敘,使用 PacBio 技術對全長 16S rRNA 進行定序,將整體提高細菌鑑定的分辨率。
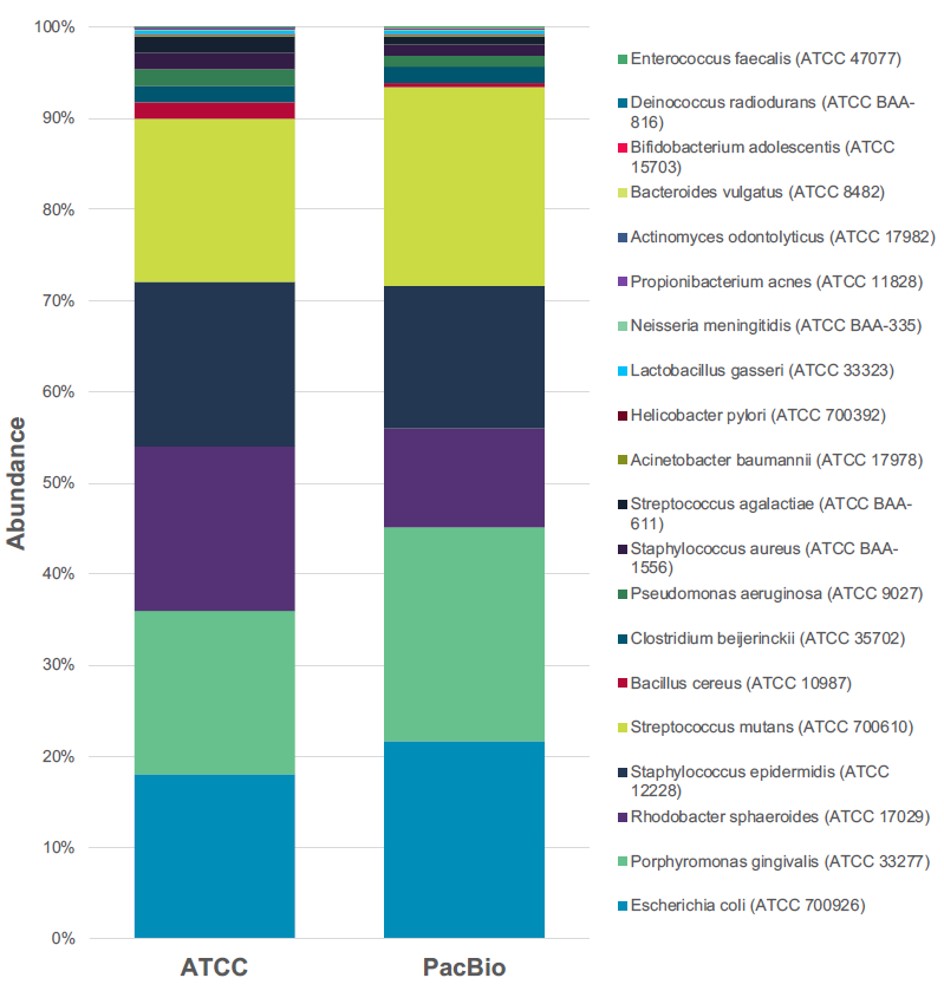
細菌研究中的另一個常見分析是總體基因體分析。總體基因體分析通常是指環境中複雜樣本,該樣本擁有多樣性生物學特徵。總體基因體分析面臨的難點是樣本中的菌種異質性、豐度不均勻和高度變化[2,3],樣品的多樣性增加了樣品中細菌定性的難度。透過第三代定序,它可以提高總體基因體學研究的分辨率。為了驗證 PacBio 定序技術在總體基因體分析中的能力,官方測試一商用細菌混樣,內含 20 株已知菌種的細菌,依照特定比例去組成的樣品 (ATCC®MSA-1003™),藉由 PacBio 三代定序的方法去做鑑定。分析結果顯示 >99% 的 16S rRNA 序列與 MSA-1003 樣品預期的組成相符 (如下圖所示)。顯示運用第三代定序技術,能真實地辨識混合物樣品中的細菌組成。
參考資料
1. Johnson, Jethro S., et al. "Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis." Nature communications 10.1 (2019): 1-11.
2. Treangen, Todd J., et al. "MetAMOS: a modular and open source metagenomic assembly and analysis pipeline." Genome biology 14.1 (2013): 1-20.
3. Schloss, Patrick D., and Jo Handelsman. "Metagenomics for studying unculturable microorganisms: cutting the Gordian knot." Genome biology 6.8 (2005): 1-4.
16S 核糖體 RNA (16S ribosomal RNA, 16S rRNA) 為原核生物核醣體小次單元的重要組成,16S rRNA 序列包含保守區以及 9 個高度變異區,稱為 V1-V9。而這些高度變異區域是具有特異性的序列,可做為細菌系統發育和分類的依據。因此,16S rRNA 基因定序已被廣泛用於鑑定各個領域的細菌研究。
全長 16S rRNA 從 V1 到 V9 通常約 1,500 個鹼基對。過去,檢測 16S rRNA 菌相的主要工具是次世代定序 (Next-generation sequencing, NGS)。由於次世代定序的讀長限制,大多數 NGS 技術只能檢測到 16S rRNA 的一部分。例如,Roche-454 定序儀檢測 V1 至 V3 區域,而 Illumina 技術通常定序 V3 和 V4 區域的序列。然而,最近研究發現,只讀一部份的 16S rRNA 去做細菌的註解,會使得某些細菌被偵測到的機率較低,進一步影響統計出來的菌相或者細菌豐度。
2019 年一篇由 Jethro S. 等人發表在 Nature 的文章中指出,進行菌相分析時,只讀取 16S rRNA 部分區域的序列,可能會使得細菌在分類上效果不好。如下左表顯示[1],當只定序 V1-V2 區域時,對於變形菌門 (Proteobacteria) 的辨識度是差的,只定序 V3-V5 區域時,對於放線菌門 (Actinobacteria) 的辨識度也不好。根據右圖物種階層熱樹圖[1],僅定序 V4 區域時,樹狀圖呈現深紅色分枝,表示未被分類到的細菌百分比很高。當 V1-V9 全部定序時,其樹狀圖顏色接近白色,表示大部分的菌都是有被分類到的。另一方面,只定序部分區域,在細菌分類學分析時,並不是一直都不適用的。例如,大腸桿菌和志賀氏菌的辨別度在只定序 V1-V3 區域時,也有良好的分辨度。總結來說,若需要做菌相分析時,進行 V1 到 V9 全區域的定序,對於微生物菌相分析時,可以提供更良好的分類結果。

第三代定序 (Third-generation sequencing, TGS) 隨著技術越來越成熟,準確度及通量上的提升,近期的研究與應用也越來越多。相較於次世代定序,可以讀取較長片段的三代定序,是作為 16S rRNA V1-V9 全區域定序的好工具。與二代定序相比,三代定序技術可以輕鬆定序超過 20 Kb,足以對全長 16S rRNA 進行定序。尤其是使用 PacBio 技術,定序可以實現長讀長、高準確度,以及平均定序等特性。
在三代定序還未被應用前,16S rRNA 部分區域定序,可以將細菌鑑定到屬的層級。根據研究顯示,分類至屬的層級時,95% 的定序準確度將導致50%的細菌被錯誤分類。而當鑑定的層級要從屬進階到種時,所需的定序準確度要求更高,應將定序準確性提高到至少 98% 以上,在這個準確度的情況下,約還有 50% 的細菌會被錯誤分類 (data from Dr. Zhu Baoli, Institute of Microbiology, CAS)。因此,要定序 16S rRNA V1-V9 全長區域,所需的技術需要讀的長且準。綜合以上所敘,使用 PacBio 技術對全長 16S rRNA 進行定序,將整體提高細菌鑑定的分辨率。
細菌研究中的另一個常見分析是總體基因體分析。總體基因體分析通常是指環境中複雜樣本,該樣本擁有多樣性生物學特徵。總體基因體分析面臨的難點是樣本中的菌種異質性、豐度不均勻和高度變化[2,3],樣品的多樣性增加了樣品中細菌定性的難度。透過第三代定序,它可以提高總體基因體學研究的分辨率。為了驗證 PacBio 定序技術在總體基因體分析中的能力,官方測試一商用細菌混樣,內含 20 株已知菌種的細菌,依照特定比例去組成的樣品 (ATCC®MSA-1003™),藉由 PacBio 三代定序的方法去做鑑定。分析結果顯示 >99% 的 16S rRNA 序列與 MSA-1003 樣品預期的組成相符 (如下圖所示)。顯示運用第三代定序技術,能真實地辨識混合物樣品中的細菌組成。

參考資料
1. Johnson, Jethro S., et al. "Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis." Nature communications 10.1 (2019): 1-11.
2. Treangen, Todd J., et al. "MetAMOS: a modular and open source metagenomic assembly and analysis pipeline." Genome biology 14.1 (2013): 1-20.
3. Schloss, Patrick D., and Jo Handelsman. "Metagenomics for studying unculturable microorganisms: cutting the Gordian knot." Genome biology 6.8 (2005): 1-4.
圖爾思生物科技 / 微生物體研究中心
許瑄珉 文案



