全基因體定序
Whole Genome Sequencing (WGS)
人類全基因體定序(WGS)可應用於疾病研究和人類演化等研究,可辨識基因體序列和結構上的變化:單核甘酸多態性 (SNP)、基因片段插入/缺失 (INDEL)、拷貝數變異 (CNV)、結構變異(SV)。
加入詢價車
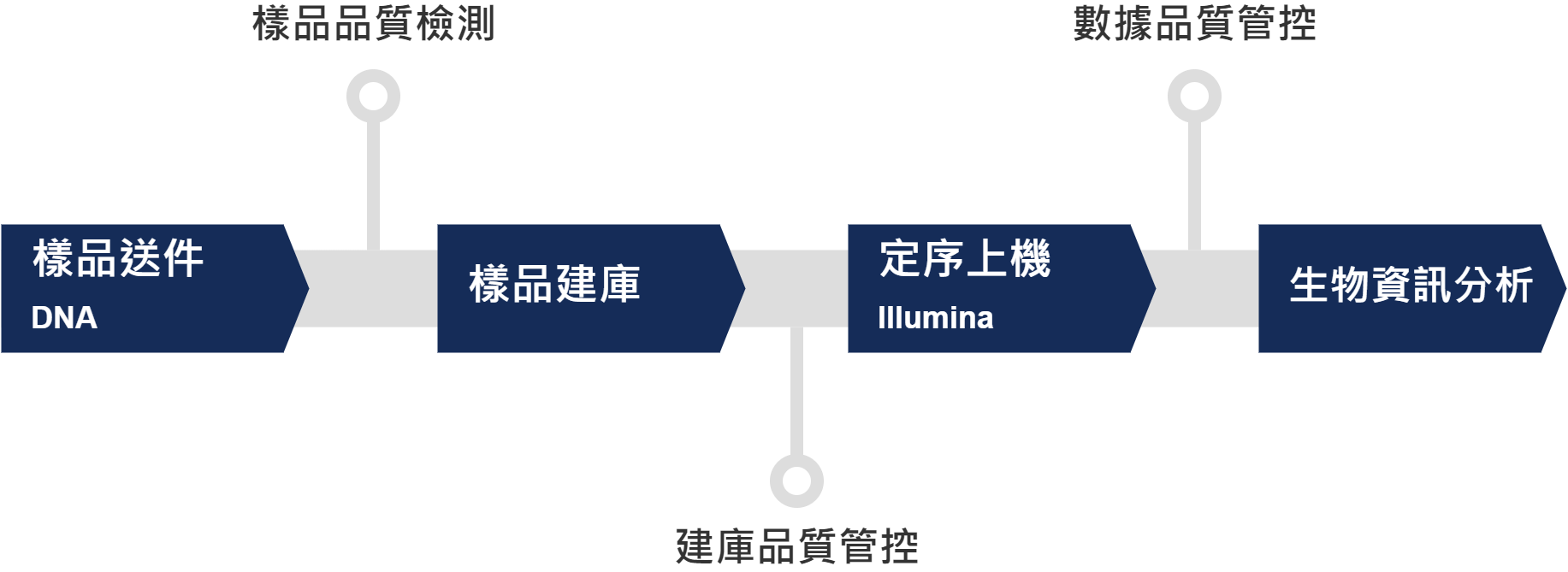
服務流程
項目週期:樣品通過檢測後,35-42 個工作天(含分析)。

分析流程
生物資訊分析流程主要分為兩部分,除了進行基本分析的基因體變異辨認(variant calling,藍色區塊),還可根據辨認的變異進行深入的進階分析 (橘色區塊,欲進行進階分析請於送件之前洽詢)。
癌症組織樣品定序可進行 Somatic 分析流程。
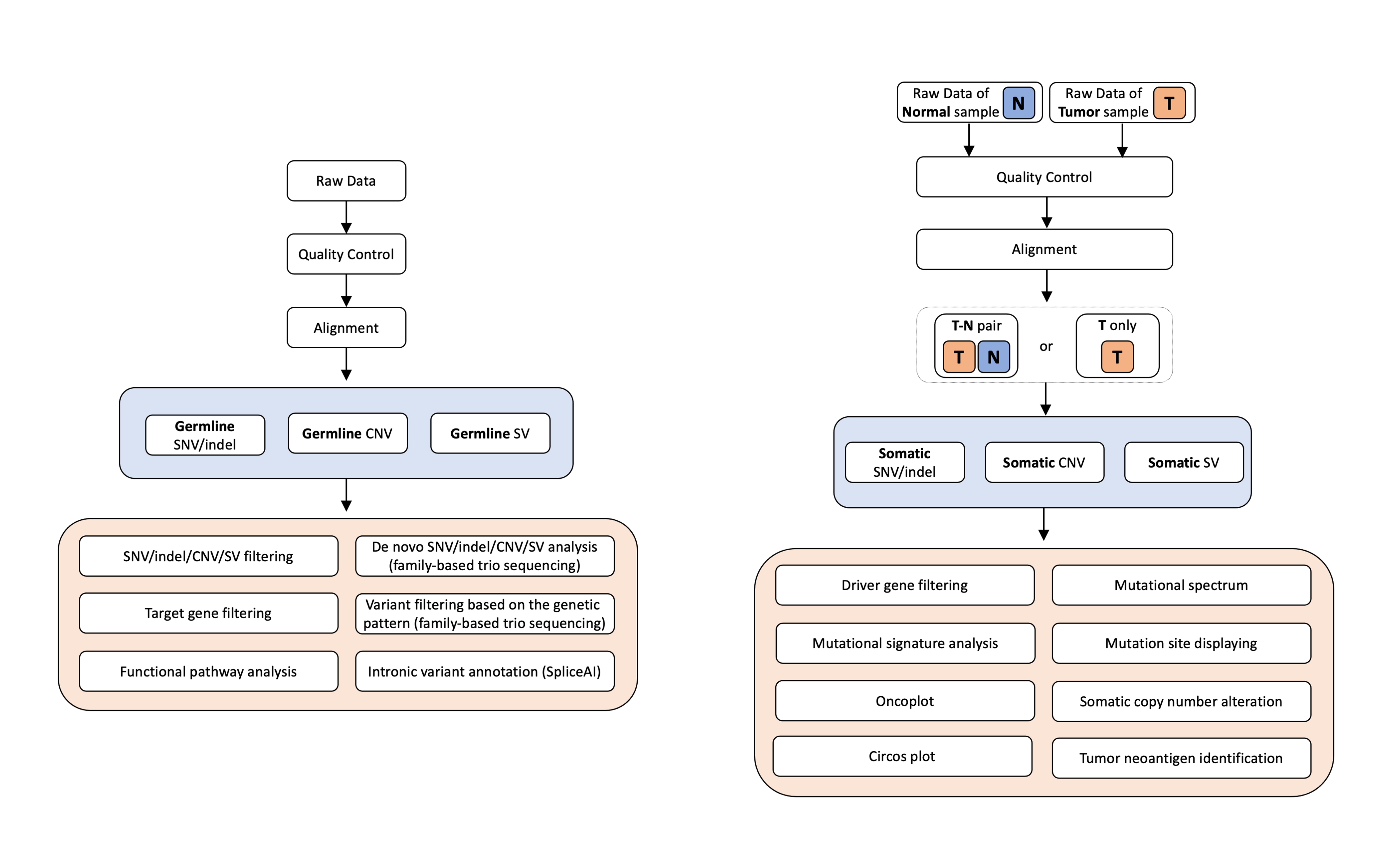
癌症組織樣品定序可進行 Somatic 分析流程。

樣品需求
DNA總量:
一般樣品: ≧ 1.0 μg
石蠟包埋樣品(FFPE sample): ≧ 1.6 μg
DNA 濃度:
≧ 50 ng/ μl (樣品濃度定量Qubit®測定為主)
樣品體積:
≧ 20 μl
樣品純度:
OD260/OD280 = 1.8-2.0
DNA無降解且無汙染
建議先行以電泳膠圖確認片段大小及DNA完整度,並於送件時提供膠圖結果
(A) DNA Marker, (B) 合格樣品 (C) 降解樣品 (D) RNA汙染樣品 (E) 蛋白質殘留汙染樣品 (以紅框標記汙染處)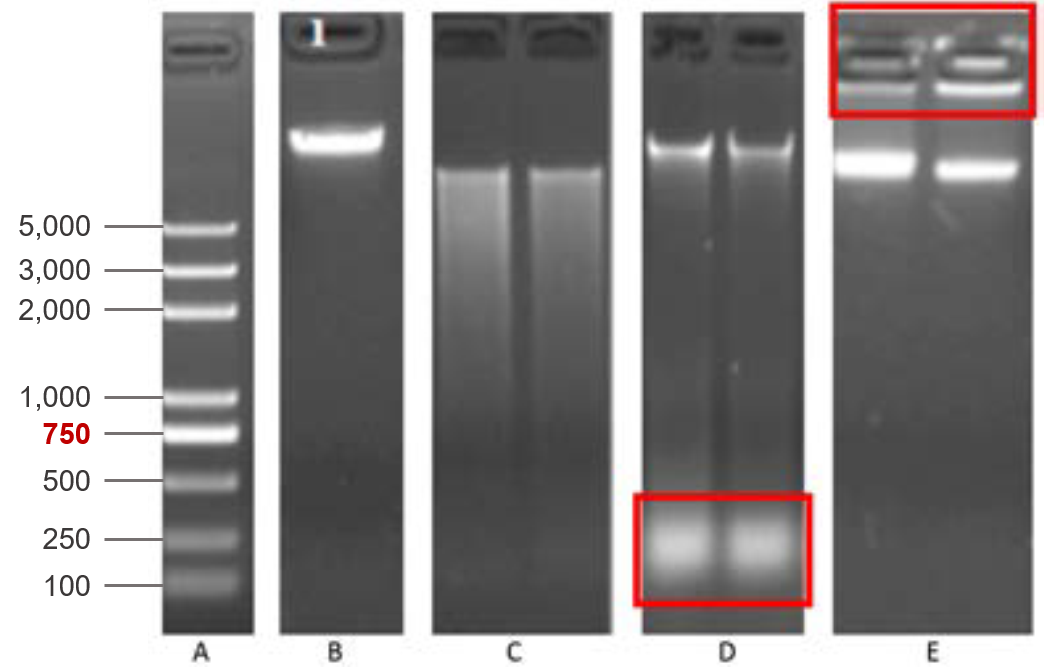
一般樣品: ≧ 1.0 μg
石蠟包埋樣品(FFPE sample): ≧ 1.6 μg
DNA 濃度:
≧ 50 ng/ μl (樣品濃度定量Qubit®測定為主)
樣品體積:
≧ 20 μl
樣品純度:
OD260/OD280 = 1.8-2.0
DNA無降解且無汙染
建議先行以電泳膠圖確認片段大小及DNA完整度,並於送件時提供膠圖結果
(A) DNA Marker, (B) 合格樣品 (C) 降解樣品 (D) RNA汙染樣品 (E) 蛋白質殘留汙染樣品 (以紅框標記汙染處)

定序規格
Novaseq 6000, paired-end 150 bp
採樣建議
l 血液:
- 新鮮/冷凍保存全血:1 mL,建議使用 3 or 5mL紫頭管,冷藏盡速送樣。
- Buffy coat :0.2 mL,-80度保存及運送。
l 組織:
- 新鮮或冷凍組織檢體:100-200 mg (米粒大小),以液態氮速凍,-80度保存及運送。。
l 細胞:
- 細胞樣本:1x10^6~1x10^7 cells,-80度保存及運送。
其他注意事項
- 全血樣品避免使用玻璃材質且需加入EDTA抗凝劑,避免反覆凍融或放置時間過長,低溫運送。
- 組織採集後立即用預冷的0.9%生理鹽水進行潤洗,並剔除非研究組織,將樣本分割成50 mg左右的小塊,立即以液態氮快速冷凍,保存管使用parafilm封口,-80℃ 保存。
- 細胞樣本準備:加細胞冷凍保存液(血清+10%DMSO),以液態氮慢凍/收集得到細胞沉澱,液態氮快速冷凍。
- 寄送時將樣本管放入更大的管或者夾鏈袋中,防止樣本運輸過程中被破壞,並用大體積乾冰運輸,保證圖爾思驗收時有足夠乾冰剩餘。
常見問題
Q1
人類全基因體定序 (WGS) 應用在哪些方面?
A1
1) 在疾病研究方面:全基因體範圍內搜尋疾病相關候選位點或區域,特別適合於有較少分子研究基礎疾病類型或研究結構變異的情況。
2) 人類演化、比較基因體學等的研究:可利用全基因體的基因資訊全面進行種族演化、種族特異性基因及區域的篩選。
2) 人類演化、比較基因體學等的研究:可利用全基因體的基因資訊全面進行種族演化、種族特異性基因及區域的篩選。
Q2
人類全基因體定序 (WGS) 的優勢為何?
A2
與外顯子定序與RNA定序技術相比,技術流程較為單純而成熟、易操作、易實現;與外顯子定序捕獲技術相比,可全面挖掘各種遺傳變異,特別是一些大的結構變異 (倒置、易位、拷貝數變異) 以及一些高GC含量的區域 (也就是較難被 WES 定序到的區域),在全基因體範圍內尋找與疾病或功能相關的位點;與傳統晶片分型技術 (例如SNP array) 相比,可發掘全新的 (novel) 和罕見 (rare) 的變異。
Q3
全基因體定序 (WGS) 一般建議多少的定序深度?
A3
定序深度需要根據研究目的及預算來制定,一般建議至少達到平均 30x 的定序深度,30x 已經能夠檢測出大部份的變異位點 (遺傳變異),但是在如果希望檢測出更多或是更大規模的結構變異,或是腫瘤組織攜帶的變異,建議可以採用 50-60x 左右的定序深度。
Q4
全基因體定序 (WGS) 是否可以定序到粒線體 DNA(mtDNA) 的序列呢?
A4
雖然 WGS 或是 WES 的定序數據中,能夠涵蓋到一小部分的粒線體序列,但是常規分析會過濾掉大多數的 mtDNA 位點,分析結果的可信度也較低,除了使用專為 mtDNA 設計的比對流程之外,也建議可以嘗試透過捕獲 mtDNA 建庫定序的方式來取得更為可信的結果。
Q5
INDEL 或 SNV 的 vcf 檔,各樣品 genotype 的欄位要如何閱讀?
A5
vcf (variant call format) 檔為記載變異資訊的檔案,每個樣品提供的註釋資訊順序,是根據欄位 FORMAT 提供的資訊排列,該欄位縮寫名稱的意思,可參考以下 vcf 檔案的說明文件 (https://samtools.github.io/hts-specs/VCFv4.2.pdf)。
Q6
全基因體定序 (WGS) 與全外顯子定序 (WES) 分析上的差異?
A6
WGS 和 WES 主要差別在於涵蓋人類基因體的區域大小不同。目前在分析 single nucleotide variation (SNV)、insertion and deletion (Indel) 和拷貝數變異 (copy number variation, CNV) 上,資料來源是WGS或是WES用的分析工具大致上是相同的,都是使用GATK (genomic analysis toolkit)的分析流程。不過在分析大片段的變異,例如:結構變異 (structural variation, SV),建議使用 WGS 的定序數據來分析。因為 WGS 的優勢在於對於基因體覆蓋度高並且具有較好的一致性,分析出來的結果當然比較全面並且可信度較高。不過,有時也會針對不同研究目的而使用不同的分析方法。WGS 與WES 的差異詳見次世代定序知識櫥窗(http://toolsbiotech.blog.fc2.com/blog-entry-122.html)。
Q7
文獻提到調高 coverage 有助於低頻率變異點的發現,以 allele frequency 評估並找尋變異位點的策略,僅針對低頻率變異位點來說,有存在變異的位點,需要有多少數量的reads來支持 (不包含normal位點) 才會比較可信?
A7
在這邊低頻率變異點指的是在腫瘤組織內出現的變異點,假設其頻率為 10%,表示平均而言要有 100 條 reads 才會有 10 條是有變異點的 reads,所以必須要先了解該次定序的深度為多少,才比較有依據來認定何謂可信的變異點。
Q8
人類參考基因體版本 GRCh38 和版本 hg19 相比之下,在基因的座標上有差異嗎? 如果有,可以做轉換跟對應嗎?
A8
GRCh38 與 hg19 在基因座標上是完全不一樣的,可以使用 liftover (https://genome.ucsc.edu/cgi-bin/hgLiftOver) 工具作轉換。版本差異可參考次世代定序知識櫥窗 (http://toolsbiotech.blog.fc2.com/blog-entry-119.html)
Q9
計算腫瘤突變負荷量 (tumor mutational burden, TMB),在 WGS/WES 不同定序深度間該如何校正?
A9
計算 TMB 時,的確會有很多因素影響,例如定序深度、變異篩選的條件、還有樣品內腫瘤細胞和正常細胞的比例…等因素都會直接影響到計算 TMB 的數值,目前似乎還沒有公認比較好的校正方式,也許在未來研究人員收集更多資訊之後會有比較好的解決方式,可參考Fancello, L., Gandini, S. et al. Tumor mutational burden quantification from targeted gene panels: major advancements and challenges. j. immunotherapy cancer 7, 183 (2019),這篇論文中有詳細的討論。
Q10
請問目前有許多不同的分析變異方法,可以互相比較嗎?或是該如何選擇?
A10
現在分析方法相當的多,也有很多文獻是進行分析方法的比較,比較內容大致有sensitivity (敏感度), Specificity (特異度), False positive rate (偽陽性率), Positive predictive value (陽性預測值), False discovery rate (錯誤發現率)等這幾項,各有其優缺點,我們所選擇的是目前最多人使用的分析變異方法,而有一些大型研究也會採用多種分析變異的方法之後採用部分聯集或是交集的方式,增加變異的正確率。
Q11
目前有哪些資料庫可以用在變異註解 (annotation)?
A11
變異註解對於研究人員相當地重要,可用在理解變異的影響,以及利用註解進行篩選,簡略介紹如下,詳細項目請參閱圖爾思提供的範例報告。
依照註解內容可以分為五大部份:
第一部分:變異位置在基因體中的區域、變異種類等,使用RefSeq、Ensembl的資料庫。
第二部分:使用不同的人群資料庫,如 dbSNP150、COSMIC、1000 genomes、ExAC、gnomAD、ClinVar 等進行比對,可了解該變異在人群中的發生頻率。
第三部份:使用多種功能預測軟體預測變異造成的可能影響,如 Polyphen、SIFT等。
第四部份:變異資訊,如:定序深度、基因型及變異比例等,此部分來源是分析時所得到的結果。
第五部份:與基因功能資料庫 (Gene Ontology)、人類疾病資料庫 (OMIM)、人類罕見疾病資料庫 (Orpha)、生物功能途徑資料庫 (KEGG, PID, BIOCARTA, REACTOME) 進行比對。
依照註解內容可以分為五大部份:
第一部分:變異位置在基因體中的區域、變異種類等,使用RefSeq、Ensembl的資料庫。
第二部分:使用不同的人群資料庫,如 dbSNP150、COSMIC、1000 genomes、ExAC、gnomAD、ClinVar 等進行比對,可了解該變異在人群中的發生頻率。
第三部份:使用多種功能預測軟體預測變異造成的可能影響,如 Polyphen、SIFT等。
第四部份:變異資訊,如:定序深度、基因型及變異比例等,此部分來源是分析時所得到的結果。
第五部份:與基因功能資料庫 (Gene Ontology)、人類疾病資料庫 (OMIM)、人類罕見疾病資料庫 (Orpha)、生物功能途徑資料庫 (KEGG, PID, BIOCARTA, REACTOME) 進行比對。
Q12
在研究癌症體細胞變異時,如果沒有正常細胞當作配對的樣品,只有癌組織的DNA,可否分析出體細胞變異呢?
A12
一般在分析體細胞變異時,最好要有同一患者的正常細胞DNA當作配對的樣品,因為這樣才能刪去遺傳性變異的部分,得到的才是屬於體細胞變異。但是如果研究上真的無法得到這樣的配對樣品,目前 GATK 有開發 tumor-only 的分析模式,利用演算法和大型人群資料庫當作篩選的方法,刪除的效果當然沒有利用配對樣品 DNA 來的好,所以分析出來的結果當中還是有部分是屬於遺傳性變異。或是另一種方式就是提供一群健康者的 (非配對) DNA 進行定序,當作對照組以幫助篩選出體細胞變異。
Q13
GATK 版本的差異?是否要將過去的分析改用新版重新分析才有可比性?
A13
不同的版本分析出來的變異數目,以 GATK3 和 GATK4 進行比較,差異蠻大,所以建議如果是同一個研究,應該採取相同的版本分析,而且不僅是尋找變異的工具要版本一致外,資料前處理的分析流程應該也要採取相同的版本和參數設定。



