26
2022.08
站在大神的肩膀: 三代定序 WGS/SV 應用重點文獻分享
原創文章 引用請註明出處
抵押所有人品!
召喚~~~神...隊友!?
世人都知道神隊友的出現,可遇不可求,有時候賭上所有人品也抽不到一張好牌 (泣
但是! 神...肩膀的存在是已知的,只需要設定目標仔細挖掘一定會獲得寶藏的
現在,閱讀本篇的看倌們請跟我們一起直達大神們的肩膀吧!
大神一號 (大神排名不分先後僅以年度區分)

大神貳號
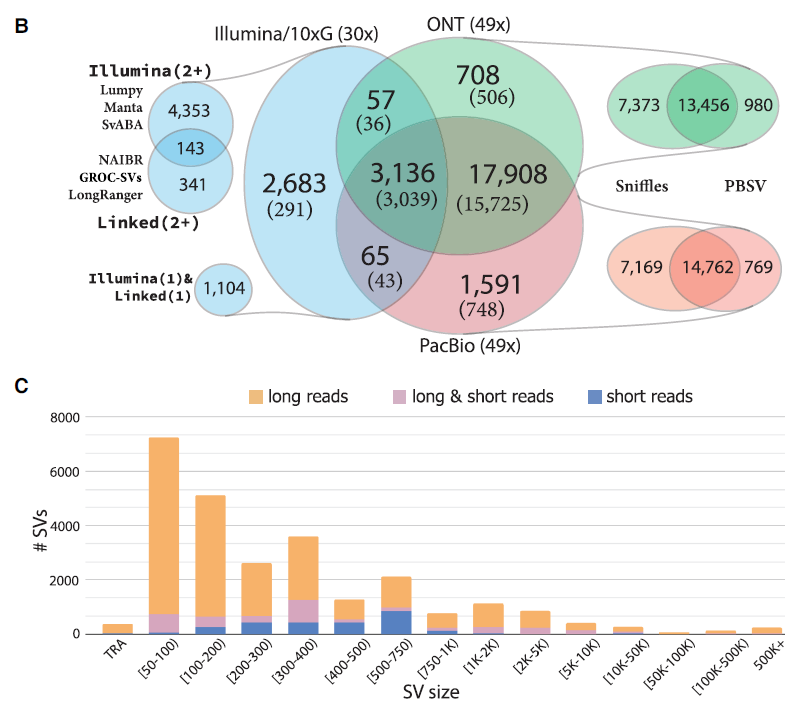
透過不同 SV caller 進行 SV 偵測,顯示三代定序發現的 SV 數量極大於 NGS 所發現的數量,且 ONT 與 PacBio 所發現的 SV 一致性高達九成。
抵押所有人品!
召喚~~~神...隊友!?
世人都知道神隊友的出現,可遇不可求,有時候賭上所有人品也抽不到一張好牌 (泣
但是! 神...肩膀的存在是已知的,只需要設定目標仔細挖掘一定會獲得寶藏的
現在,閱讀本篇的看倌們請跟我們一起直達大神們的肩膀吧!
大神一號 (大神排名不分先後僅以年度區分)

期刊: Nature
年份: 2020
定序平台: Nanopore MinION
定須規格: 155Gb / 50X
【Nature 完整端到端X染色體組裝】
2001年,首次發布人類基因體序列,經歷了20年的努力,目前最完整的人類基因體參考資料庫為GRCh38。但是,就在2020年,揭開歷史即將被改寫的序幕!
今天要介紹的文獻利用Nanopore長讀長定序,組裝出完整人類X染色體,從端粒(telomere)到端粒,重建了著絲粒阿爾法衛星陣列(centromeric satellite DNA array),並且填補過去文獻中X染色體上的29個缺口,其中包含了假染色體區域(human pseudoautosomal regions)和癌症-睾丸擴增基因家族(cancer-testis ampliconic gene families, CT-X and GAGE)。除此之外,三代定序可直接偵測鹼基修飾的特性,使得研究能夠直接看複雜重複序列以及衛星陣列區域的甲基化情況。
人類全基因體定序共獲得155 Gb數據、平均深度為50倍,一半以上的序列長70 kb或更長,最長的讀長達1.04 Mb。一致性的準確度從組裝後的99.46%,經由Nanopore與先前的PacBio數據拋光,準確度可達99.9%。初步組裝後的X染色體仍有三個部分待補齊,主要是著絲粒及大片段重複區域,利用著絲粒中已知的衛星重複序列去進行『定位』,搭配超長讀長的Nanopore技術,成功解讀遺失的資訊,完整定序出人類X染色體序列。
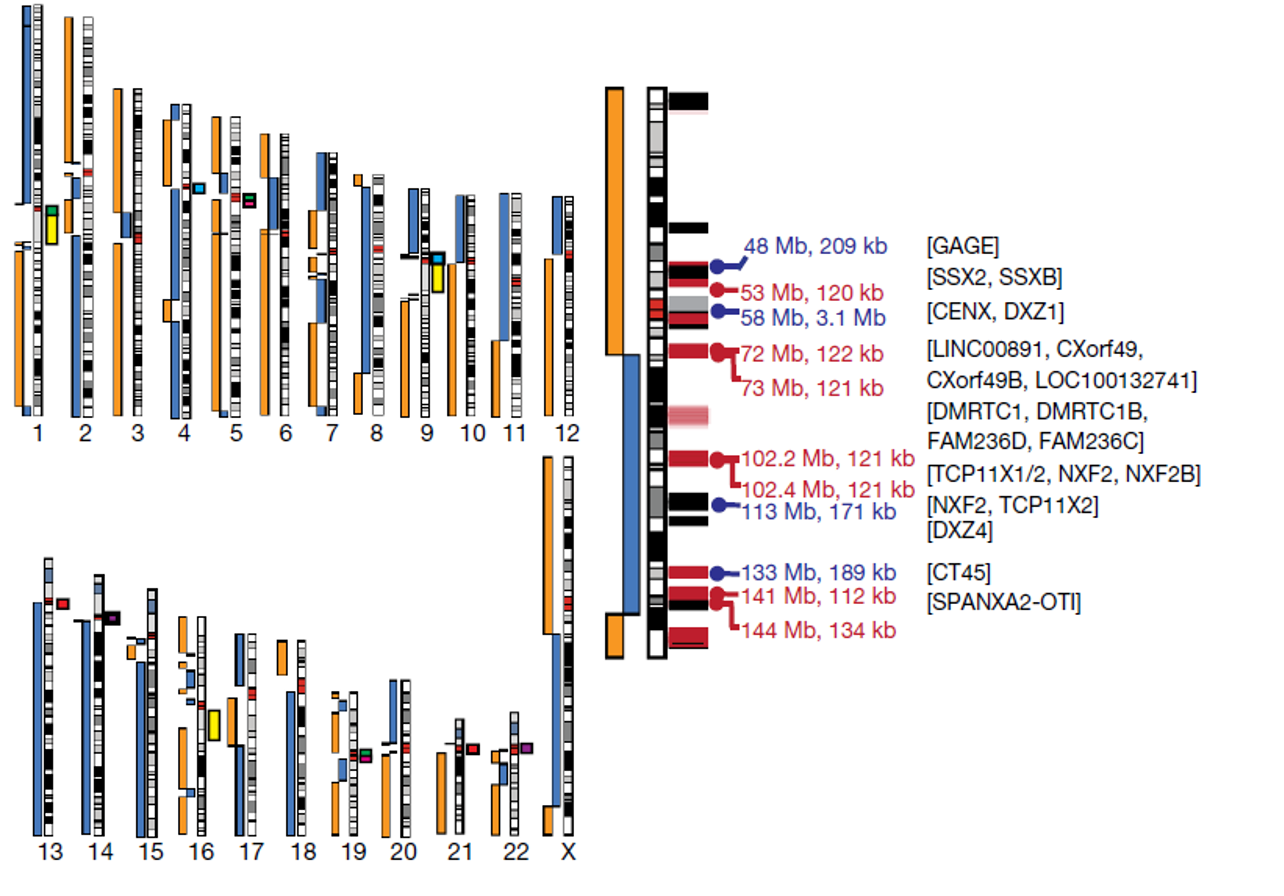
藍色及橘色長方表示無Gap的contig,大部分斷裂處皆位於中心粒區域,其餘色塊顯示了預期在非同源染色體之間序列相似的著絲粒衛星陣列:chr1、chr5 和 chr19(綠色);chr4 和 chr9(淺藍色); chr5 和 chr19(粉紅色); chr13 和 chr21(紅色); 和 chr14 和 chr22(紫色)。

藍色及橘色長方表示無Gap的contig,大部分斷裂處皆位於中心粒區域,其餘色塊顯示了預期在非同源染色體之間序列相似的著絲粒衛星陣列:chr1、chr5 和 chr19(綠色);chr4 和 chr9(淺藍色); chr5 和 chr19(粉紅色); chr13 和 chr21(紅色); 和 chr14 和 chr22(紫色)。
主要成就:
- 完整組裝端粒到端粒
- 填補過去參考基因體所有29個Gap
ONT 優勢:
- 單分子讀長達1.04 Mb
- 在組裝上有兩個複雜斷點是藉由 Nanopore超長片段搭配 PacBio 先前數據填補而成。
大神貳號

期刊: Genome Research
年份: 2020
定序技術: PacBio, Nanopore
定須規格: 81~177Gb / 23X-49X
【Genome Research_Nanopore, PacBio及Illumina/10x Genomics於乳腺癌中SV的偵測】
目前在使用短讀長定序進行癌症相關研究中面臨到的一個主要限制,即為短讀長定序系統地遺漏了許多重要變異,根據先前文獻指出短讀長定序分析後所檢測出之結構變異(SVs)由於系統性錯誤,偽陽性及偽陰性的比例高達50%。對SV偵測的疏漏可能使其對整個類別的遺傳危險因素及其參與的相關癌症進展和患者存活率視而不見。改善對癌症中SV的識別可以提供更有針對性和更有效的治療選擇,並增進我們對疾病及其進展的基本了解。
研究團隊使用Illumina / 10x Genomics,Pacific Biosciences (PacBio)和Oxford Nanopore Technologies (ONT) 對SKBR3乳腺癌細胞株以及源自兩名乳腺癌患者的腫瘤和正常組織進行了全基因體定序。定序結果使用多個SV call sets推斷SV和大規模特異性等位基因的拷貝數變異(CNV)。研究表明長讀長定序可顯著提高SV檢測的準確度和靈敏度,其中>90%的SV可同時在PacBio與ONT偵測,顯示出兩個平台對SV偵測的高度一致性。另外,即使在相對較低的覆蓋範圍(25×-30×)下,PacBio與ONT皆可達召回率>80%且準確率>90%之結果。本篇研究指出在乳腺癌患者的腫瘤和正常組織中偵測到之SV一致性高達77% (20,388/26,148),顯示出患者對特定參考基因體的需求。最後作者將SV和CNV數據整合到統一的核型圖結構中,以更準確地呈現突變的癌症基因體,發現已知癌症相關基因中的數百種變異只能通過長讀長定序才能檢測到。凸顯了對癌症基因體進行長讀長定序以準確分析其基因不穩定性的必要性。

透過不同 SV caller 進行 SV 偵測,顯示三代定序發現的 SV 數量極大於 NGS 所發現的數量,且 ONT 與 PacBio 所發現的 SV 一致性高達九成。
主要成就:
- 建立癌症大規模變異檢測方法
- 確立三代定序技術平台間SV檢測一致性
ONT 優勢:
- 提升SV發現準確度與靈敏度
- 低覆蓋率下(>24X),召回率>80%; 精確度>90%
- 與PacBio平台發現之SV一致性高達9成
大神參號

期刊: Nature
年份: 2021
定序技術: Nanopore, PacBio, 10X Chromium, Bionano, Hi-C. Illumina
定須規格:
以蜂鳥為代表
- PacBio / 77Gb / 69X
- ONT / 29Gb / 18X
- Bionano / 838Gb / 751X
- Hi-C / 272Gb / 243X
- Illumina / 173Gb / 159X
- 10X Chromium / 56Gb / 50X
【脊椎動物基因體計劃】
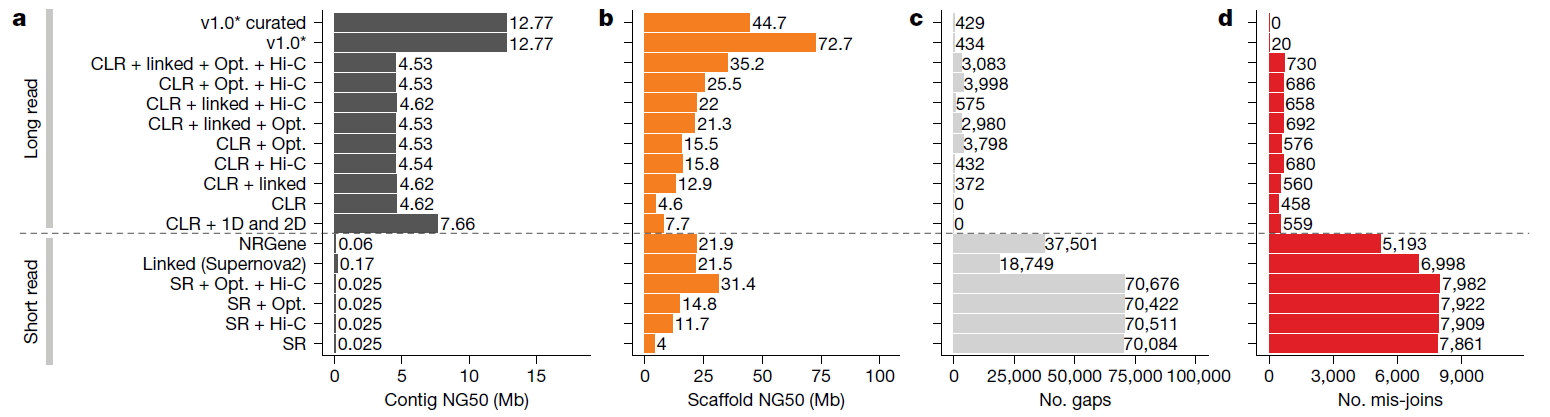
本篇由脊椎動物基因體計劃(VGP)所發表,涵蓋了6個主要脊椎動物種系中的16個物種的基因體組裝結果。研究團隊首先選擇基因體較小(約1Gb)且已具二代定序數據註解之參考基因體的物種Anna’s 蜂鳥(Calypte anna)進行多種定序技術及組裝方法的廣泛評估以測試出最佳的組裝策略,組裝結果發現PacBio CLR或ONT數據比Illumina數據長30~300倍;在contig NG50的數據顯示短讀長結果約為0.025~0.169 Mb,而長讀長結果達4.6~7.66 Mb,Gap也由18,000~70,000個(二代定序數據),減少到約400~4,000個(三代定序數據),且組裝結果比較無關數據類型組合或使用的演算法,證明,在當前的定序技術和演算法的情況下,僅通過短讀長技術不可能實現高連續性的Contig,因為無法跨越長於讀長的重複序列區段。然後,使用蜂鳥基因體組裝得出的最佳化公式構建了具有單倍型分型的CLR contigs的VGP組裝流程(v1.0),結合10x Genomics、Bionano與Hi-C進行骨架組裝、缺口填補,鹼基校正,最後由人工調整。系統性的測試了剩下15個物種的基因體組裝。組裝結果校正了先前基因體中上千個基因的重大錯誤,針對高GC區段的調控以及染色體的演化等也有進一步的發現,確認三代定序技術對於獲得最佳化基因體組裝至關重要,如果處理不當,未解決的複雜重複片段和單倍型雜合體會是裝配錯誤的主要來源。本篇研究結果奠定了後續的基因註解和基因體功能分析的基礎。
主要成就:
- 單篇完成了16個物種基因體組裝
- 包含性染色體皆組裝良好
- 校正先前參考基因體組裝錯誤
- 確立脊椎動物組裝策略
ONT 優勢:
- contig NG50提升45~184倍至7.66Mb
- 減少17~45倍的Gap數量
- 提供單倍型資訊
大神肆號

期刊: Genome Medicine
年份: 2021
定序技術: Nanopore
定須規格: 53.9 Gb/17X
【揭露人類複雜結構和結構變異的起源】
人類基因體中存在大量遺傳變異和體細胞突變。遺傳變異與個體之間的疾病風險和表型變異有關 。眾所周知,體細胞突變會導致癌症發生和罕見疾病 。鑑定這些變異和突變是人類遺傳學中的一個關鍵問題。
在癌症基因體定序研究中,對 SV 的分析對於發現驅動基因和癌症發生機制也很重要。然而,我們對體細胞 SVs 的理解仍然不完整,體細胞組織中 SVs 的產生機制仍然難以捉摸。
為了全面了解種系變異和體細胞突變,並推斷 SV 產生的生物學機制,研究團隊使用 ONT 對 11 個日本肝癌和匹配的正常樣本進行了全基因體定序。這些樣本先前已被國際癌症基因體聯盟 (ICGC) 定序。
在 polymorphic germline SVs 中,分析結果確定了 8,004 個插入、6,389 個缺失、27 個倒位和 32 個染色體內易位,且插入和刪除事件之間的模式也不同。在插入事件中,發現大多數插入的來源是由 transposons 和 Alu 引起,而其他類型的插入,例如串聯重複和加工假基因,則很少見。而缺失則是由幾種機制產生的,非同源末端連接 (NHEJ)、替代末端連接 (alt-EJ)、非等位基因同源重組 (NAHR)、folk stalling 和模板轉換或微同源介導的斷裂誘導修復 (FoSTeS/MMBIR),研究團隊推斷缺失產生的機制,發現大多數 NAHR 事件是由 SINE 中的重組錯誤引起的。
本篇研究結果闡明了長讀長在分析人類多態性和體細胞突變方面的優勢。但由於短讀長具有較低的定序錯誤率,可將長讀長和短讀長結合起來可以揭露更完整的體細胞突變和種系變異。
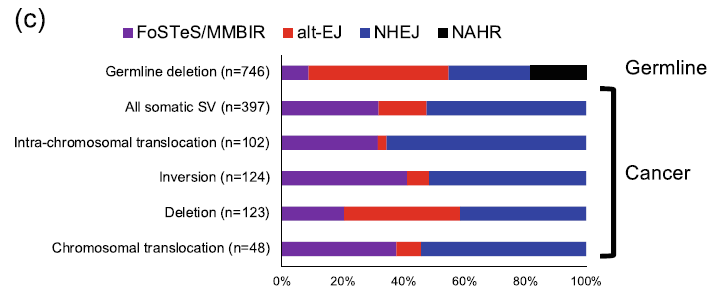
肝癌樣本中,不同 SV 可能的形成機制,在癌症體細胞中的結構變異形成機制顯著不同於生殖細胞

肝癌樣本中,不同 SV 可能的形成機制,在癌症體細胞中的結構變異形成機制顯著不同於生殖細胞
主要成就:
- 提供人類遺傳與癌症的結構變異與形成起源的全面目錄
ONT 優勢:
- 由於reads長度較長,大多數reads可被映射到唯一的人類基因體,並且具更大比例的reads可用於variant calling;
- Reads 比重複元件長,可以檢測到由重複類型組成之 SV;
- 低覆蓋深度(長讀長 17 倍與短讀長約 40 倍)仍然檢測出較高體細胞 SV 數量(增加了 1.6 倍);
- 可以分析插入的完整序列,能夠檢測插入的transposable elements和處理過的假基因的結構;
- 可以觀察到體細胞 SVs 的單倍型結構;
- 露SV 的真實結構,由repeat elements引起的 SV 在短讀長可能被判定為不同類型的 SV;
- 除了遺傳變異和體細胞突變之外,還可以做甲基化分析
最後也歡迎對三代定序有興趣或想取得相關研究領域優先門票的研究大神們可以參考這裡,挑選合適的研究神器喔!
參考資料
- Miga KH, Koren S, Rhie A, Vollger MR, Gershman A, Bzikadze A, Brooks S, Howe E, Porubsky D, Logsdon GA, Schneider VA, Potapova T, Wood J, Chow W, Armstrong J, Fredrickson J, Pak E, Tigyi K, Kremitzki M, Markovic C, Maduro V, Dutra A, Bouffard GG, Chang AM, Hansen NF, Wilfert AB, Thibaud-Nissen F, Schmitt AD, Belton JM, Selvaraj S, Dennis MY, Soto DC, Sahasrabudhe R, Kaya G, Quick J, Loman NJ, Holmes N, Loose M, Surti U, Risques RA, Graves Lindsay TA, Fulton R, Hall I, Paten B, Howe K, Timp W, Young A, Mullikin JC, Pevzner PA, Gerton JL, Sullivan BA, Eichler EE, Phillippy AM. Telomere-to-telomere assembly of a complete human X chromosome. Nature. 2020 Sep;585(7823):79-84. doi: 10.1038/s41586-020-2547-7. Epub 2020 Jul 14. PMID: 32663838; PMCID: PMC7484160.
- Aganezov S, Goodwin S, Sherman RM, Sedlazeck FJ, Arun G, Bhatia S, Lee I, Kirsche M, Wappel R, Kramer M, Kostroff K, Spector DL, Timp W, McCombie WR, Schatz MC. Comprehensive analysis of structural variants in breast cancer genomes using single-molecule sequencing. Genome Res. 2020 Sep;30(9):1258-1273. doi: 10.1101/gr.260497.119. Epub 2020 Sep 4. PMID: 32887686; PMCID: PMC7545150.
- Rhie A, McCarthy SA, Fedrigo O, Damas J, Formenti G, Koren S, Uliano-Silva M, Chow W, Fungtammasan A, Kim J, Lee C, Ko BJ, Chaisson M, Gedman GL, Cantin LJ, Thibaud-Nissen F, Haggerty L, Bista I, Smith M, Haase B, Mountcastle J, Winkler S, Paez S, Howard J, Vernes SC, Lama TM, Grutzner F, Warren WC, Balakrishnan CN, Burt D, George JM, Biegler MT, Iorns D, Digby A, Eason D, Robertson B, Edwards T, Wilkinson M, Turner G, Meyer A, Kautt AF, Franchini P, Detrich HW 3rd, Svardal H, Wagner M, Naylor GJP, Pippel M, Malinsky M, Mooney M, Simbirsky M, Hannigan BT, Pesout T, Houck M, Misuraca A, Kingan SB, Hall R, Kronenberg Z, Sović I, Dunn C, Ning Z, Hastie A, Lee J, Selvaraj S, Green RE, Putnam NH, Gut I, Ghurye J, Garrison E, Sims Y, Collins J, Pelan S, Torrance J, Tracey A, Wood J, Dagnew RE, Guan D, London SE, Clayton DF, Mello CV, Friedrich SR, Lovell PV, Osipova E, Al-Ajli FO, Secomandi S, Kim H, Theofanopoulou C, Hiller M, Zhou Y, Harris RS, Makova KD, Medvedev P, Hoffman J, Masterson P, Clark K, Martin F, Howe K, Flicek P, Walenz BP, Kwak W, Clawson H, Diekhans M, Nassar L, Paten B, Kraus RHS, Crawford AJ, Gilbert MTP, Zhang G, Venkatesh B, Murphy RW, Koepfli KP, Shapiro B, Johnson WE, Di Palma F, Marques-Bonet T, Teeling EC, Warnow T, Graves JM, Ryder OA, Haussler D, O'Brien SJ, Korlach J, Lewin HA, Howe K, Myers EW, Durbin R, Phillippy AM, Jarvis ED. Towards complete and error-free genome assemblies of all vertebrate species. Nature. 2021 Apr;592(7856):737-746. doi: 10.1038/s41586-021-03451-0. Epub 2021 Apr 28. PMID: 33911273; PMCID: PMC8081667.
- Fujimoto A, Wong JH, Yoshii Y, Akiyama S, Tanaka A, Yagi H, Shigemizu D, Nakagawa H, Mizokami M, Shimada M. Whole-genome sequencing with long reads reveals complex structure and origin of structural variation in human genetic variations and somatic mutations in cancer. Genome Med. 2021 Apr 29;13(1):65. doi: 10.1186/s13073-021-00883-1. PMID: 33910608; PMCID: PMC8082928.
圖爾思生物科技 / 微生物體研究中心
吳雁韻 文案
http://www.toolsbiotech.com/
© BIOTOOLS. All Rights Reserved



