24
2021.12
【平台大亂鬥:比較二代與三代平台之間的再現性、準確性和實用性 (上)】
原創文章 引用請註明出處
隨著定序成本的降低,高通量DNA定序技術在臨床檢測與基礎生醫研究中已被視為常用方法,其用途包含但不限於個體或族群間的變異偵測、基因體物種等級的鑑定,辨識混合物中的物種多樣性等[1,2]。DNA定序可能會因各種因素影響上機結果,包含樣品收集、建庫的準備、定序機型及試劑,以及後續的生物資訊分析手法,以上各項都會影響DNA定序的準確性及覆蓋率。在過去,微陣列晶片品質控制聯盟 (The Microarray Quality Control Consortium) 曾做過多項大型研究,特別去看 RNA-seq 的再現性、 RNA-seq 的品質控制、 RNA-seq 與微陣列晶片之間的再現性、數據處理及標準化的最佳方式[3-5]。然而,卻沒有類似研究探討DNA定序在不同重要平台之間的再現性。本文要跟大家介紹的文獻就是比較二代與三代各平台之間的再現性、準確性和實用性[6]。因文獻內容較多,故分為上、下集,上集會針對定序品質、序列對應率、覆蓋率、錯誤率去探討,下集則討論單核苷酸與插入/缺失異常、結構變異偵測率、細菌定序結果。時間緊迫者可直接拉至文末觀看上集結論。
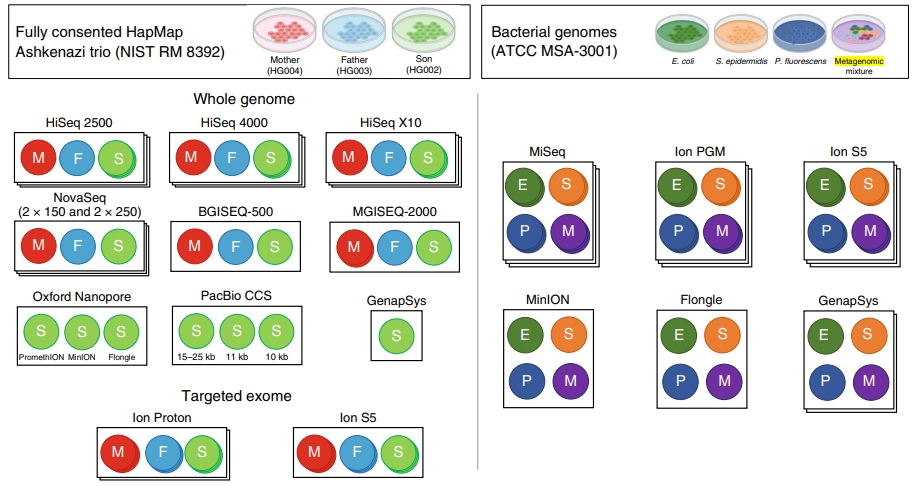
【研究背景介紹】 本篇研究比較多平台於不同實驗室間定序人類基因體、三隻菌株,以及含 10 種菌的混合物總基因體定序結果。人類樣品從個人基因體計畫 (Personal Genome Project, PGP) 建立而來的 RM8392 細胞株,為德系猶太人家族檢體,分別是母親 (HG004)、父親 (HG003)、兒子 (HG002)。單菌檢體從美國典型培養物保藏中心 (American Type Culture Collection, ATCC) 而來,總基因體細菌檢體則包含不同GC含量、革蘭氏染色、生態學和生理學的細菌混合於單一樣品中,編號 ATCC MSA-3001。每個機台與相對應的定序樣品標示如圖一。
定序平台則包含:
二代定序平台
Illumina (HiSeq 4000, HiSeq X10, NovaSeq 6000, MiSeq)
ThermoFisher Scientific (Ion PM/S5)
BGI (BGISEQ-500/MGISEQ-2000)
GenapSys GS111
三代定序平台
Oxford Nanopore Technologies (Flongle/MinION/PromethION)
Pacific Biosciences (CCS mode)
【圖一】
【數據品質比較】
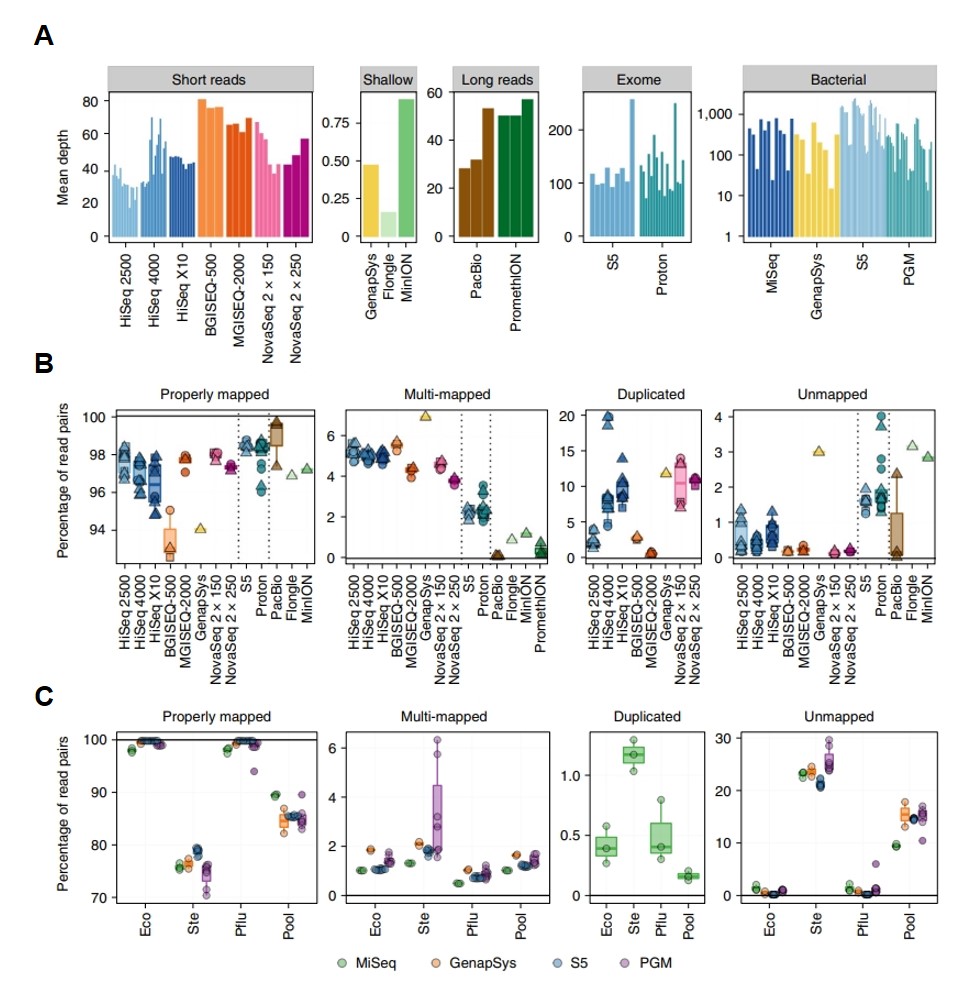
定序深度隨著定序平台及樣品有所差異,從細菌定序平均深度落於 1000X 至淺覆蓋率概括整個基因體 (平均覆蓋率 <1X ),如圖 2A,大部分的全基因體平均深度落在 25X - 80X。整體來說,鹼基質量分數、 GC 分佈、平衡序列含量、低 N 值和低重複序列數等指標,顯示出所有定序數據的品質為好的。而序列對應率 (mapping rate) 在同一機型內的一致性為高的,但是在機器間的一致性則差異很大,如圖 2B。BGISEQ-500 和 GenapSys GS111 擁有最低的短讀長特有對應率 (short-read unique mapping efficiency)、最高的多重對應率 (multi-mapping rate)。 ThermoFisher 的對應率略優於 Illumina 及 MGI。三代定序PacBio的對應率擇優於 Nanopore,Nanopore 的對應率遠低於其他平台,落於 85% 左右。
單菌及細菌總基因體的部分僅使用 Illumina、Ion Torrent、Nanopore、GenapSys 等平台進行定序。總基因體中的細菌基因體大小、 GC 比例等特質在各平台之間會影響被偵測到的機率。其對應率也被發現直接與該種菌的序列有關,對應率在不同種菌之間的差異很大,而不同平台間的一致性則為高的,如圖 2C。
【圖二】
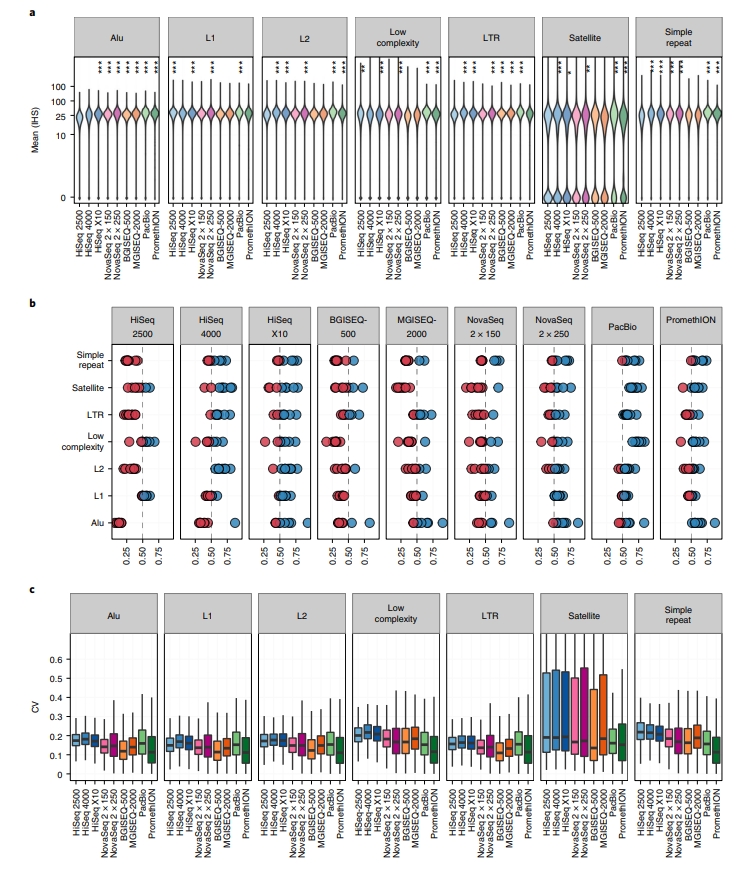
【覆蓋率比較】 接著去看各平台在全基因體某些特定區域的覆蓋率,像是 Alu、L1、L2、長串聯重複片段 (Long Tandem Repeats, LTRs)、低複雜度區域、衛星序列、重複序列等區域。不同平台間,不管是二代定序或者三代定序,各區域間覆蓋率的分布一致性是高的,如圖 3A。可以發現 HiSeq 2500、BGISEQ-500、MGISEQ-2000 在多個區域有較低的覆蓋率,但是 HiSeq 2500 在低複雜度區域有較好的覆蓋率,而 BGISEQ-500、MGISEQ-2000 在 Alu 區域有較好的覆蓋率。HiSeq 4000、HiSeq X10 在 L2、長串聯重複片段及簡單重複序列區域有較好的覆蓋率。然而,還是有特定平台在所有區域中,其覆蓋率是優於其他平台平均值的。以PacBio 及 Nanopore PromethION 來說,這兩個平台在所有區域的覆蓋率上優於其他平台。依據不同平台或者不同區域統整出的結果可見圖 3B-C。
【圖三】
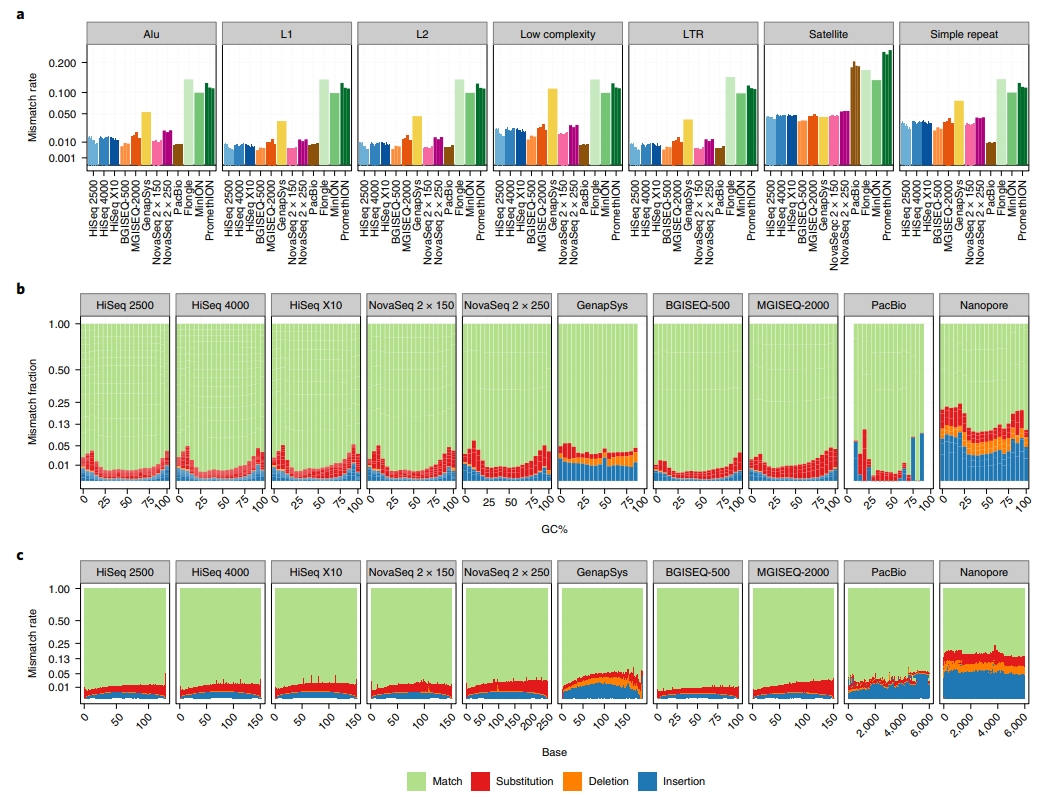
【與參考序列間的錯誤配對率比較】 為了評估定序結果與參考序列間的配對率,各平台數據與UCSC RepeatMasker 區域進行比對,已獲得錯誤配對率 (mismatch rate),見圖 4A。整體來說,和 Nanopore 平台比起來,二代定序平台擁有較低的錯誤配對率,而 PacBio 平台與二代定序平台比起來擁有差不多,甚至是較低的錯誤配對率,特別是在衛星區域有較佳的表現。以二代定序平台來說,BGISEQ-500 比起 HiSeq 2500、HiSeq 4000、HiSeq X10 擁有更低的錯誤配對率,而 GS111 除了在衛星區域的錯誤配對率較高外,其他區域的錯誤配對率皆比剩餘的二代定序平台佳。值得一提的是,NovaSeq 2×250-bp 比起 2×150-bp 試劑來說,擁有更低的錯誤配對率,而錯誤配對率也會受GC百分比比例及每個鹼基在讀長中的位置而影響 (圖 4B-C)。以GC百分比比例來看,大部分平台在 GC 比例小於 25 或者是大於 75 時,會有較多的替換、插入/刪除發生。而所有二代定序、PacBio 平台在定序時,靠近 3 端的序列會有錯誤率增加的現象,Nanopore平台不管是哪一機型,在同一定序片段上其錯誤率相同。
【圖四】
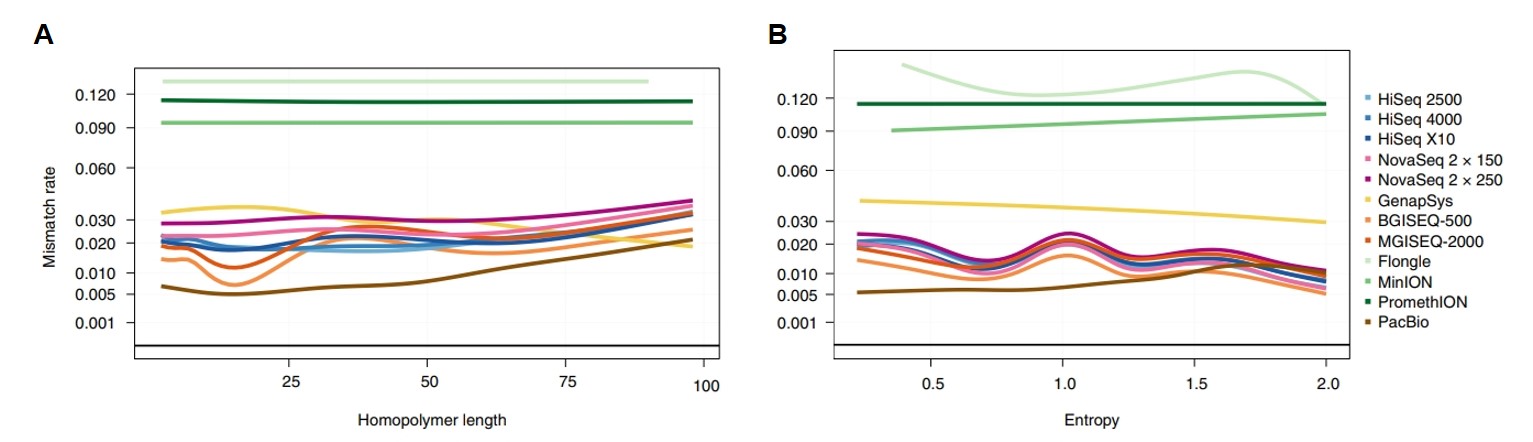
接著根據 UCSC Table Browser 將各平台讀取片段依據 Tandem Repeat Finder 的定義去做分層,分同元序列 (homopolymers) 和短串聯重複片段 (short tandem repeats, STRs) 去看其定序效益 (圖 5A-B)。不管是在同元序列或是短片段串聯重複序列中,PacBio 皆擁有最低的錯誤配對率。而二代定序平台中,BGISEQ-500、MGISEQ-2000 在短的同元序列中表現比 Illumina 平台優,GenapSys GS111 則有最差的表現。以大於25 bp的長同元序列來比較,所有二代定序平台有著差不多的定序效益。
 【圖五】
【圖五】
平台大亂鬥:比較二代與三代平台之間的再現性、準確性和實用性 (上) 的介紹就到此為止。最後總結一下本篇文獻針對目前已經介紹的幾個指標給的建議,包含以下幾點:
(1) 序列對應效率 (mapping rate) 取決於定序的機型和樣品物種。以二代定序平台來說,Illumina 有著最好的表現,而 BGISEQ-500 和 GenapSys GS111 因定序片段較短,有最低的短讀長特有對應率和最高的多重對應率。三代定序平台 PacBio 擁有最高的特有對應率和最低的未對應率。
(2) BAM檔可以利用 mosdepth 進行平均體染色體覆蓋率的計算以達到標準化,接著使用 Picard DownsampleSam 進行減少取樣 (down sampling)。不過,就算數據已經進行標準化,在同一儀器中,不同區域像是重複序列、低複雜度區域可能還是會有不均一的覆蓋率。所有平台中,長讀長的三代定序,可以提供基因體序列的最高覆蓋率。而二代定序平台中則以 HiSeq 4000、HiSeq X10 有最一致且高的覆蓋率。
(3) 定序錯誤 (sequencing error) 可以利用 BBMap reformat.sh 來計算,並且與 mismatch histogram tables 做比較。所有的平台皆有定序錯誤率,從小至 0.1%、 大至衛星序列區域的 20%。所有二代定序平台中, BGI/MGISEQ 提供最低的定序錯誤率,PacBio 為所有平台中擁有最低定序錯誤率的機型, Nanopore 則為所有平台中擁有最高定序錯誤率的平台。然而, Nanopore不管是最小的 Flongle 或者是最大的 PromethION,其定序錯誤率之間是高度一致的。
(4) 錯誤配對率 (mismatch rate) 在低 GC 或者高 GC 區域會有提升的現象。而在大片段重複序列、同元序列或者是短片段串聯重複序列等區域,會有更頻繁的錯誤。其中,以 PacBio CCS 模式擁有最低的錯誤率,而 Nanopore 平台的錯誤率最高,且不依區域不同而有所差異。
參考資料
1. DePristo, Mark A., et al. "A framework for variation discovery and genotyping using next-generation DNA sequencing data." Nature genetics 43.5 (2011): 491-498.
2. MacLean, Daniel, Jonathan DG Jones, and David J. Studholme. "Application of'next-generation'sequencing technologies to microbial genetics." Nature Reviews Microbiology 7.4 (2009): 96-97.
3. Maqc Consortium. "The MicroArray Quality Control (MAQC) project shows inter-and intraplatform reproducibility of gene expression measurements." Nature biotechnology 24.9 (2006): 1151.
4. Shi, Leming, et al. "The MicroArray Quality Control (MAQC)-II study of common practices for the development and validation of microarray-based predictive models." Nature biotechnology 28.8 (2010): 827-838.
5. Li, Sheng, et al. "Multi-platform assessment of transcriptome profiling using RNA-seq in the ABRF next-generation sequencing study." Nature biotechnology 32.9 (2014): 915-925.
6. Foox, Jonathan, et al. "Performance assessment of DNA sequencing platforms in the ABRF Next-Generation Sequencing Study." Nature Biotechnology 39.9 (2021): 1129-1140.
隨著定序成本的降低,高通量DNA定序技術在臨床檢測與基礎生醫研究中已被視為常用方法,其用途包含但不限於個體或族群間的變異偵測、基因體物種等級的鑑定,辨識混合物中的物種多樣性等[1,2]。DNA定序可能會因各種因素影響上機結果,包含樣品收集、建庫的準備、定序機型及試劑,以及後續的生物資訊分析手法,以上各項都會影響DNA定序的準確性及覆蓋率。在過去,微陣列晶片品質控制聯盟 (The Microarray Quality Control Consortium) 曾做過多項大型研究,特別去看 RNA-seq 的再現性、 RNA-seq 的品質控制、 RNA-seq 與微陣列晶片之間的再現性、數據處理及標準化的最佳方式[3-5]。然而,卻沒有類似研究探討DNA定序在不同重要平台之間的再現性。本文要跟大家介紹的文獻就是比較二代與三代各平台之間的再現性、準確性和實用性[6]。因文獻內容較多,故分為上、下集,上集會針對定序品質、序列對應率、覆蓋率、錯誤率去探討,下集則討論單核苷酸與插入/缺失異常、結構變異偵測率、細菌定序結果。時間緊迫者可直接拉至文末觀看上集結論。
【研究背景介紹】 本篇研究比較多平台於不同實驗室間定序人類基因體、三隻菌株,以及含 10 種菌的混合物總基因體定序結果。人類樣品從個人基因體計畫 (Personal Genome Project, PGP) 建立而來的 RM8392 細胞株,為德系猶太人家族檢體,分別是母親 (HG004)、父親 (HG003)、兒子 (HG002)。單菌檢體從美國典型培養物保藏中心 (American Type Culture Collection, ATCC) 而來,總基因體細菌檢體則包含不同GC含量、革蘭氏染色、生態學和生理學的細菌混合於單一樣品中,編號 ATCC MSA-3001。每個機台與相對應的定序樣品標示如圖一。
定序平台則包含:
二代定序平台
Illumina (HiSeq 4000, HiSeq X10, NovaSeq 6000, MiSeq)
ThermoFisher Scientific (Ion PM/S5)
BGI (BGISEQ-500/MGISEQ-2000)
GenapSys GS111
三代定序平台
Oxford Nanopore Technologies (Flongle/MinION/PromethION)
Pacific Biosciences (CCS mode)

【圖一】
【數據品質比較】
定序深度隨著定序平台及樣品有所差異,從細菌定序平均深度落於 1000X 至淺覆蓋率概括整個基因體 (平均覆蓋率 <1X ),如圖 2A,大部分的全基因體平均深度落在 25X - 80X。整體來說,鹼基質量分數、 GC 分佈、平衡序列含量、低 N 值和低重複序列數等指標,顯示出所有定序數據的品質為好的。而序列對應率 (mapping rate) 在同一機型內的一致性為高的,但是在機器間的一致性則差異很大,如圖 2B。BGISEQ-500 和 GenapSys GS111 擁有最低的短讀長特有對應率 (short-read unique mapping efficiency)、最高的多重對應率 (multi-mapping rate)。 ThermoFisher 的對應率略優於 Illumina 及 MGI。三代定序PacBio的對應率擇優於 Nanopore,Nanopore 的對應率遠低於其他平台,落於 85% 左右。
單菌及細菌總基因體的部分僅使用 Illumina、Ion Torrent、Nanopore、GenapSys 等平台進行定序。總基因體中的細菌基因體大小、 GC 比例等特質在各平台之間會影響被偵測到的機率。其對應率也被發現直接與該種菌的序列有關,對應率在不同種菌之間的差異很大,而不同平台間的一致性則為高的,如圖 2C。

【圖二】
【覆蓋率比較】 接著去看各平台在全基因體某些特定區域的覆蓋率,像是 Alu、L1、L2、長串聯重複片段 (Long Tandem Repeats, LTRs)、低複雜度區域、衛星序列、重複序列等區域。不同平台間,不管是二代定序或者三代定序,各區域間覆蓋率的分布一致性是高的,如圖 3A。可以發現 HiSeq 2500、BGISEQ-500、MGISEQ-2000 在多個區域有較低的覆蓋率,但是 HiSeq 2500 在低複雜度區域有較好的覆蓋率,而 BGISEQ-500、MGISEQ-2000 在 Alu 區域有較好的覆蓋率。HiSeq 4000、HiSeq X10 在 L2、長串聯重複片段及簡單重複序列區域有較好的覆蓋率。然而,還是有特定平台在所有區域中,其覆蓋率是優於其他平台平均值的。以PacBio 及 Nanopore PromethION 來說,這兩個平台在所有區域的覆蓋率上優於其他平台。依據不同平台或者不同區域統整出的結果可見圖 3B-C。

【圖三】
【與參考序列間的錯誤配對率比較】 為了評估定序結果與參考序列間的配對率,各平台數據與UCSC RepeatMasker 區域進行比對,已獲得錯誤配對率 (mismatch rate),見圖 4A。整體來說,和 Nanopore 平台比起來,二代定序平台擁有較低的錯誤配對率,而 PacBio 平台與二代定序平台比起來擁有差不多,甚至是較低的錯誤配對率,特別是在衛星區域有較佳的表現。以二代定序平台來說,BGISEQ-500 比起 HiSeq 2500、HiSeq 4000、HiSeq X10 擁有更低的錯誤配對率,而 GS111 除了在衛星區域的錯誤配對率較高外,其他區域的錯誤配對率皆比剩餘的二代定序平台佳。值得一提的是,NovaSeq 2×250-bp 比起 2×150-bp 試劑來說,擁有更低的錯誤配對率,而錯誤配對率也會受GC百分比比例及每個鹼基在讀長中的位置而影響 (圖 4B-C)。以GC百分比比例來看,大部分平台在 GC 比例小於 25 或者是大於 75 時,會有較多的替換、插入/刪除發生。而所有二代定序、PacBio 平台在定序時,靠近 3 端的序列會有錯誤率增加的現象,Nanopore平台不管是哪一機型,在同一定序片段上其錯誤率相同。

【圖四】
接著根據 UCSC Table Browser 將各平台讀取片段依據 Tandem Repeat Finder 的定義去做分層,分同元序列 (homopolymers) 和短串聯重複片段 (short tandem repeats, STRs) 去看其定序效益 (圖 5A-B)。不管是在同元序列或是短片段串聯重複序列中,PacBio 皆擁有最低的錯誤配對率。而二代定序平台中,BGISEQ-500、MGISEQ-2000 在短的同元序列中表現比 Illumina 平台優,GenapSys GS111 則有最差的表現。以大於25 bp的長同元序列來比較,所有二代定序平台有著差不多的定序效益。
 【圖五】
【圖五】平台大亂鬥:比較二代與三代平台之間的再現性、準確性和實用性 (上) 的介紹就到此為止。最後總結一下本篇文獻針對目前已經介紹的幾個指標給的建議,包含以下幾點:
(1) 序列對應效率 (mapping rate) 取決於定序的機型和樣品物種。以二代定序平台來說,Illumina 有著最好的表現,而 BGISEQ-500 和 GenapSys GS111 因定序片段較短,有最低的短讀長特有對應率和最高的多重對應率。三代定序平台 PacBio 擁有最高的特有對應率和最低的未對應率。
(2) BAM檔可以利用 mosdepth 進行平均體染色體覆蓋率的計算以達到標準化,接著使用 Picard DownsampleSam 進行減少取樣 (down sampling)。不過,就算數據已經進行標準化,在同一儀器中,不同區域像是重複序列、低複雜度區域可能還是會有不均一的覆蓋率。所有平台中,長讀長的三代定序,可以提供基因體序列的最高覆蓋率。而二代定序平台中則以 HiSeq 4000、HiSeq X10 有最一致且高的覆蓋率。
(3) 定序錯誤 (sequencing error) 可以利用 BBMap reformat.sh 來計算,並且與 mismatch histogram tables 做比較。所有的平台皆有定序錯誤率,從小至 0.1%、 大至衛星序列區域的 20%。所有二代定序平台中, BGI/MGISEQ 提供最低的定序錯誤率,PacBio 為所有平台中擁有最低定序錯誤率的機型, Nanopore 則為所有平台中擁有最高定序錯誤率的平台。然而, Nanopore不管是最小的 Flongle 或者是最大的 PromethION,其定序錯誤率之間是高度一致的。
(4) 錯誤配對率 (mismatch rate) 在低 GC 或者高 GC 區域會有提升的現象。而在大片段重複序列、同元序列或者是短片段串聯重複序列等區域,會有更頻繁的錯誤。其中,以 PacBio CCS 模式擁有最低的錯誤率,而 Nanopore 平台的錯誤率最高,且不依區域不同而有所差異。
參考資料
1. DePristo, Mark A., et al. "A framework for variation discovery and genotyping using next-generation DNA sequencing data." Nature genetics 43.5 (2011): 491-498.
2. MacLean, Daniel, Jonathan DG Jones, and David J. Studholme. "Application of'next-generation'sequencing technologies to microbial genetics." Nature Reviews Microbiology 7.4 (2009): 96-97.
3. Maqc Consortium. "The MicroArray Quality Control (MAQC) project shows inter-and intraplatform reproducibility of gene expression measurements." Nature biotechnology 24.9 (2006): 1151.
4. Shi, Leming, et al. "The MicroArray Quality Control (MAQC)-II study of common practices for the development and validation of microarray-based predictive models." Nature biotechnology 28.8 (2010): 827-838.
5. Li, Sheng, et al. "Multi-platform assessment of transcriptome profiling using RNA-seq in the ABRF next-generation sequencing study." Nature biotechnology 32.9 (2014): 915-925.
6. Foox, Jonathan, et al. "Performance assessment of DNA sequencing platforms in the ABRF Next-Generation Sequencing Study." Nature Biotechnology 39.9 (2021): 1129-1140.
圖爾思生物科技 / 微生物體研究中心
許瑄珉 文案



